
 Accessori Letto/Lettino
Accessori Letto/Lettino
Dispositivi elettromedicali: classificazione e normativa CE
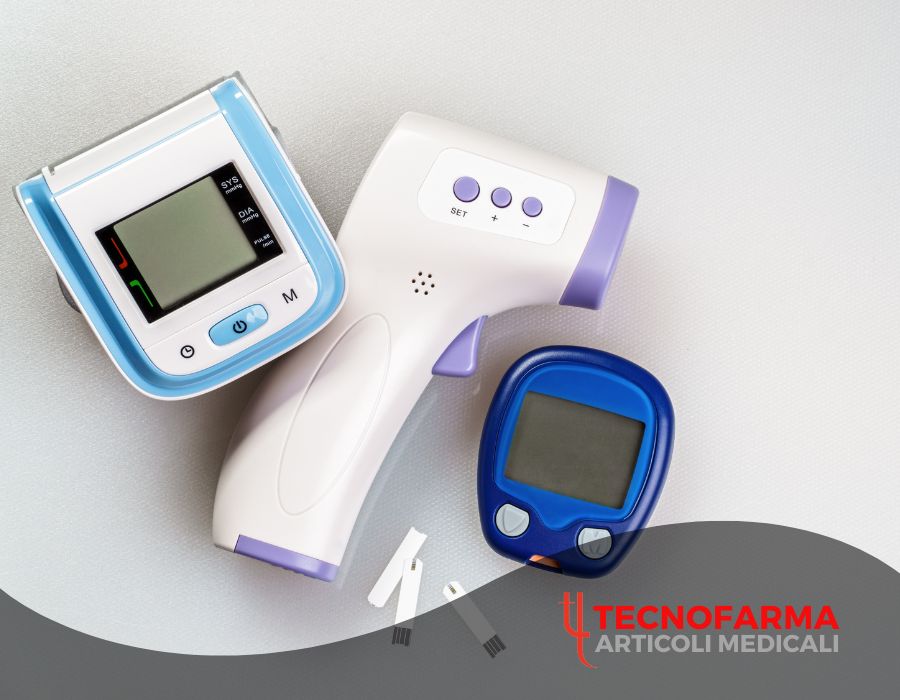
I dispositivi elettromedicali rappresentano una categoria fondamentale nell’ambito sanitario moderno, essenziali per diagnosi, monitoraggio e terapia. Comprendere la loro classificazione e gli obblighi normativi è cruciale per strutture sanitarie, professionisti della salute e distributori specializzati.
1. Cosa sono i dispositivi elettromedicali
I dispositivi elettromedicali sono apparecchiature che utilizzano energia elettrica per svolgere funzioni mediche specifiche. A differenza delle apparecchiature elettriche generiche, questi strumenti sono progettati e certificati per l’uso su pazienti o in contesti clinici.
Caratteristiche distintive:
- Alimentazione elettrica (rete o batteria)
- Destinazione d’uso medico dichiarata dal fabbricante
- Contatto diretto o indiretto con il paziente
- Conformità a standard tecnici specifici (IEC 60601)
- Marcatura CE obbligatoria per commercializzazione in UE
Esempi comuni includono elettrocardiografi, monitor multiparametrici, elettrobisturi, apparecchi per aerosolterapia, dispositivi di ventilazione, defibrillatori e apparecchiature diagnostiche per immagini. La loro presenza è ubiquitaria: dagli ospedali agli studi medici, dalle RSA alle farmacie.
2. Classificazione per classi di rischio
La normativa europea classifica i dispositivi medici, inclusi quelli elettromedicali, in quattro classi di rischio crescente. Questa classificazione determina l’iter di certificazione e gli obblighi di sorveglianza post-commercializzazione.
Classe I (rischio basso)
Dispositivi con il minor livello di rischio per il paziente, solitamente non invasivi e utilizzati per brevi periodi.
Esempi: letti ospedalieri elettrici, lampade diagnostiche, bilance medicali elettroniche, alcuni dispositivi per aerosolterapia.
Certificazione: autocertificazione del fabbricante con documentazione tecnica, senza necessità di Organismo Notificato (tranne per dispositivi sterili o con funzione di misura).
Classe IIa (rischio medio-basso)
Dispositivi che comportano un rischio moderato, spesso utilizzati per monitoraggio di parametri vitali non critici o per terapie a basso rischio.
Esempi: elettrocardiografi, monitor della pressione arteriosa, apparecchi per ultrasuoni diagnostici, pompe per infusione a gravità.
Certificazione: valutazione obbligatoria da parte di un Organismo Notificato per la conformità del sistema qualità e della documentazione tecnica.
Classe IIb (rischio medio-alto)
Dispositivi destinati a funzioni critiche di monitoraggio o terapia, il cui malfunzionamento potrebbe causare danni significativi al paziente.
Esempi: ventilatori polmonari, monitor multiparametrici per terapia intensiva, apparecchi per dialisi, dispositivi per radioterapia.
Certificazione: controllo rigoroso da Organismo Notificato, che include valutazione del progetto, del sistema qualità e della documentazione clinica.
Classe III (rischio alto)
Dispositivi a più alto rischio, essenziali per il supporto vitale o che influenzano funzioni fisiologiche critiche.
Esempi: pacemaker, defibrillatori impiantabili, dispositivi per angioplastica, sistemi di circolazione extracorporea.
Certificazione: procedura più stringente con valutazione completa del progetto, analisi clinica approfondita e audit periodici da parte dell’Organismo Notificato.
Box informativo: come identificare la classe
La classe di rischio è indicata nella documentazione tecnica del dispositivo e nell’etichetta o nelle istruzioni d’uso. Il numero identificativo dell’Organismo Notificato (quattro cifre) accanto al marchio CE indica dispositivi di Classe IIa, IIb o III.
3. Normativa europea MDR 2017/745
Dal 26 maggio 2021 è pienamente applicabile il Regolamento (UE) 2017/745 sui dispositivi medici (MDR – Medical Device Regulation), che ha sostituito le precedenti Direttive 93/42/CEE.
Principali novità introdotte:
- Requisiti più stringenti per la documentazione clinica e la valutazione delle evidenze
- Maggiore trasparenza attraverso la banca dati europea EUDAMED
- Sorveglianza post-commercializzazione rafforzata con obbligo di report periodici
- Tracciabilità completa attraverso il sistema UDI (Unique Device Identification)
- Classificazione rivista per alcuni dispositivi software e dispositivi riutilizzabili
- Persona responsabile della conformità reglementare obbligatoria per i fabbricanti
Il MDR ha introdotto una netta distinzione tra dispositivi medici e dispositivi medico-diagnostici in vitro (IVD), regolamentati dal Regolamento (UE) 2017/746.
Impatto operativo per le strutture sanitarie:
La transizione al MDR ha comportato una revisione delle certificazioni esistenti. Molti dispositivi hanno richiesto una nuova valutazione di conformità, con possibili interruzioni temporanee nella disponibilità di alcuni prodotti. Le strutture sanitarie devono verificare che i dispositivi acquisiti mantengano certificazioni valide secondo il nuovo regolamento.
4. Obblighi per acquisto e utilizzo
L’acquisto e l’utilizzo di dispositivi elettromedicali comportano responsabilità precise per le strutture sanitarie e i professionisti.
Fase di acquisto
- Verifica della marcatura CE e del numero di Organismo Notificato
- Controllo della documentazione (dichiarazione di conformità, istruzioni d’uso in italiano)
- Validazione della destinazione d’uso rispetto alle esigenze cliniche
- Registrazione nel repertorio dispositivi medici del Ministero della Salute
- Verifica dei requisiti di installazione (elettrici, ambientali, strutturali)
Responsabilità dell’utilizzatore
Le strutture sanitarie e i professionisti che utilizzano dispositivi elettromedicali devono:
- Garantire la formazione adeguata del personale utilizzatore
- Effettuare la registrazione presso il Ministero della Salute per dispositivi di Classe IIa, IIb e III
- Nominare un responsabile della gestione dei dispositivi medici
- Implementare un sistema di vigilanza per segnalare incidenti o malfunzionamenti
- Conservare la documentazione tecnica per tutta la vita del dispositivo
- Rispettare le indicazioni del fabbricante per uso e manutenzione
Box informativo: sistema di vigilanza
Incidenti gravi o malfunzionamenti devono essere segnalati entro 10 giorni al Ministero della Salute e al fabbricante. La segnalazione è obbligatoria per strutture sanitarie e professionisti.
5. Manutenzione e calibrazione
La manutenzione programmata e la calibrazione periodica sono fondamentali per garantire sicurezza, affidabilità e prestazioni dei dispositivi elettromedicali nel tempo.
Programma di manutenzione
Manutenzione preventiva:
- Ispezioni periodiche secondo le indicazioni del fabbricante
- Verifica delle sicurezze elettriche (norme CEI 62-148)
- Controllo dei parametri operativi
- Sostituzione componenti soggetti a usura
Manutenzione correttiva:
- Intervento tempestivo in caso di guasti o anomalie
- Utilizzo esclusivo di ricambi originali o equivalenti certificati
- Esecuzione da personale tecnico qualificato
Calibrazione e controlli di qualità
La calibrazione garantisce che le misurazioni fornite dal dispositivo siano accurate e affidabili. È obbligatoria per:
- Dispositivi con funzione di misura (es. sfigmomanometri, termometri clinici)
- Apparecchiature diagnostiche che influenzano decisioni cliniche
- Dispositivi terapeutici dove il dosaggio deve essere preciso
Frequenza: definita dal fabbricante, generalmente annuale o biennale, oppure dopo riparazioni significative.
Documentazione: ogni intervento deve essere registrato nel “libretto di impianto” o sistema equivalente, indicando data, operazioni eseguite, esito e operatore.
Gestione fine vita
I dispositivi elettromedicali devono essere smaltiti secondo le normative RAEE (Rifiuti da Apparecchiature Elettriche ed Elettroniche), evitando dispersione nell’ambiente di componenti dannosi.
6. Prodotti Tecnofarma certificati
Tecnofarma si distingue nel panorama della distribuzione italiana per un catalogo completo di dispositivi elettromedicali certificati secondo il Regolamento MDR 2017/745, garantendo massima conformità e sicurezza.
Gamma di dispositivi disponibili
Diagnostica e monitoraggio:
- Elettrocardiografi portatili e da studio (Classe IIa)
- Monitor multiparametrici per terapia intensiva (Classe IIb)
- Pulsossimetri e saturimetri (Classe IIa)
- Sfigmomanometri elettronici validati clinicamente (Classe IIa)
- Termometri clinici digitali a infrarossi (Classe I con funzione di misura)
Terapia respiratoria:
- Aerosol a pistone e a ultrasuoni (Classe IIa)
- Concentratori di ossigeno per ossigenoterapia domiciliare (Classe IIb)
- Aspiratori chirurgici e mucolitici (Classe IIa)
Riabilitazione ed elettroterapia:
- Elettrostimolatori TENS per terapia del dolore (Classe IIa)
- Apparecchi per magnetoterapia (Classe IIa)
- Ultrasuoni terapeutici (Classe IIa)
Servizi a valore aggiunto
Tecnofarma non si limita alla fornitura, ma offre un supporto completo:
- Consulenza pre-acquisto per identificare il dispositivo più adatto alle esigenze specifiche
- Formazione certificata per il personale sanitario sull’uso corretto dei dispositivi
- Assistenza post-vendita con tecnici specializzati per installazione e collaudo
- Contratti di manutenzione programmata con calibrazioni periodiche certificate
- Gestione documentale per conformità normativa e tracciabilità
- Aggiornamento normativo continuo sui cambiamenti del quadro regolatorio
Tutti i dispositivi commercializzati da Tecnofarma sono accompagnati da completa documentazione in italiano, dichiarazione di conformità CE e garanzia del fabbricante, assicurando piena tracciabilità e conformità agli standard più elevati.
Schema riassuntivo: classificazione dispositivi elettromedicali
| Classe | Livello di rischio | Esempi | Certificazione |
|---|---|---|---|
| I | Basso | Letti elettrici, lampade diagnostiche, aerosol base | Autocertificazione |
| IIa | Medio-Basso | ECG, sfigmomanometri, monitor pressione | Organismo Notificato |
| IIb | Medio-Alto | Ventilatori, monitor terapia intensiva, dialisi | Organismo Notificato + audit |
| III | Alto | Pacemaker, defibrillatori impiantabili | Valutazione completa + audit periodici |
Il nostro consiglio
La classificazione dei dispositivi elettromedicali e la conformità alla normativa CE non sono semplici adempimenti burocratici, ma garanzie essenziali di sicurezza per pazienti e operatori. Affidarsi a distributori qualificati come Tecnofarma significa accedere a prodotti certificati, supporto tecnico specializzato e aggiornamento continuo sulle evoluzioni normative, elementi fondamentali per una gestione ottimale del parco tecnologico sanitario.
